
Genomics Glossary
base the building blocks of the DNA molecule. Changes at single bases can identify individual differences.
gene expression the production of a protein from the DNA blueprint.
mapping/aligning Using a reference genome to piece together the sequenced fragments of other individuals.
protein-coding gene a section of DNA that is the blueprint for a protein. Proteins are large molecules that carry out lots of different functions in an organism, especially in the cells.
read the sequence of bases from a section of DNA.
regulatory element part of the DNA that switches gene expression on or off.
scaffold a section of an assembled genome consisting of short sequences and gaps.
sequencing methods that determine the order of the bases in DNA. Short-read sequencing uses small fragments and produces a short sequence of bases, long-read sequencing can produce the sequence of bases in much longer sections of DNA.
variant – differences in the genome between two individuals of the same species. A causal variant is one that results in differences in a trait between individuals
An improved reference genome for European ash
by Daniel Wood, Postdoctoral Research Associate, RBG Kew
What is a reference genome?
A reference genome is a representation of the complete genomic sequence from a single individual. A gold-standard reference genome would contain the ordered sequence of nucleotide bases (A, C, G and T) for each of an organism’s chromosomes, with no gaps. In practice, this is very hard to achieve and costs a lot of time and money. A complete human genome sequence, including sex chromosomes, was only published in August 2023. Nevertheless, even an incomplete reference genome can be very useful.

Newly budding ash leaves
Why do we need reference genomes?
Why do scientists go to all this effort to obtain a genome sequence for just one individual? There are two main reasons:
Firstly, as most of the genome sequence for different individuals of a species is the same, insights from a single genome can broadly apply to the whole species. For example, protein-coding genes can be predicted from a reference genome to give insights into the biology of a species. The reference sequence can also be compared to reference sequences from other species – differences, such as the number of genes with a particular function, may provide insights into biological differences between the species.
The second reason, is that it enables the genomic study of many individuals within a species, at a fraction of the cost of producing the reference genome itself. Genetic differences between individuals can explain important within-species trait differences, such as disease resistance. Short-read sequencing, in which fragments of a few hundred base pairs are sequenced, can be performed cost effectively for hundreds of individuals. Whilst identifying variable sites within these fragments provides some insights, not knowing their location in the genome limits the usefulness of the study.
By mapping the fragments to a reference genome, we can identify whether the variable regions cluster together or are spread throughout the genome. Moreover, we can identify nearby genes that might underlie trait differences of interest – a variant associated with interesting biological variation could be in a gene itself, or in a sequence far away that regulates expression of that gene, or simply be genetically linked to a causal variant (variants nearby on a chromosome are more likely to be inherited together than those far away). Producing a reference genome is often a crucial first step in performing genomic research in an organism.

Short read data
Why do we need an updated reference genome for European ash?
A reference genome for Fraxinus excelsior was first published in 2016, and has subsequently been used to provide insights into ash biology. For example, comparing the reference genome to other species revealed large families of genes in the ash genome that are associated with disease resistance in other plants, potentially providing targets for tree breeding. A subsequent study in 2019 used short-read sequences from hundreds of individuals mapped to this reference genome to identify genetic variants associated with resistance to ash dieback disease, a critical threat to European ash. Although it has facilitated substantial improvements in our knowledge of ash biology, this original reference genome is not perfect – rather than having 23 scaffolds corresponding to the 23 chromosomes found in ash, the assembly is fragmented into 89,514 scaffolds. The gaps between these scaffolds could contain additional genes or regulatory elements, and closing the gaps would improve our understanding of how genes and regions of interest are linked to each other along the chromosomes.
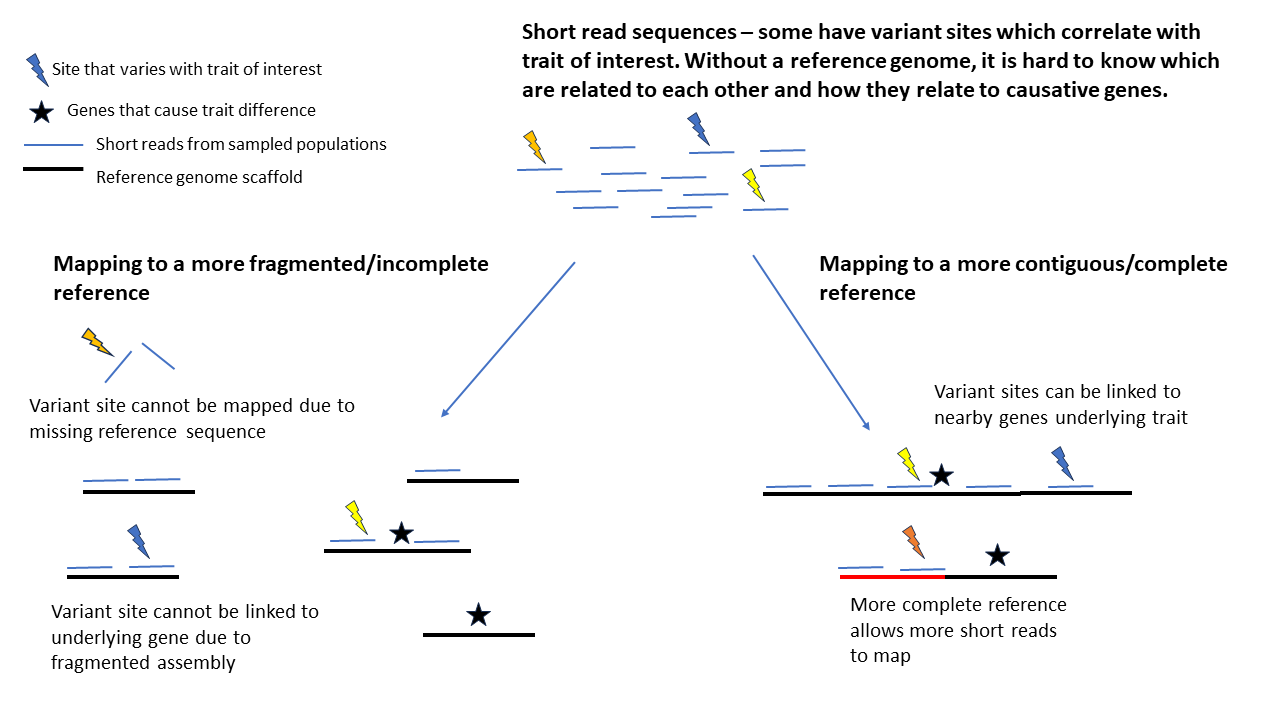
Mapping short-read sequences to fragmented compared with more complete reference genomes
What allows us to update the reference genome now?
Technological improvements are helping to close the gaps in genome assemblies. The primary drivers of these gaps are repetitive elements – where the same sequences are repeated over and over again throughout the genome. Longer repetitive elements require longer sequencing reads to bridge from the unique sequence on one side of a repeat to the unique sequence on the other. If repetitive elements are longer than the read lengths, it can be very difficult to identify the correct order of sequences within these regions. It is like trying to assemble a jigsaw puzzle with large patches of featureless blue sky between clouds – if the pieces are very small, you could probably assemble the individual clouds but would struggle to connect them together. If you had larger pieces, you could bridge the gaps between clouds and assemble a more complete picture.
Long-read sequencing technology, can now produce sequence reads that are thousands of base pairs long, rather than the hundreds of base pairs from earlier methods. These can be complemented by new methods such as HiC which capitalize on the fact that even very distant regions on the same chromosome can cluster closely together in space in the cell when the DNA is folded. The falling costs of these technologies now make it possible to use them in a wider range of organisms than ever before.
How much of an improvement is the updated reference genome?
A graft from the same individual used to produce the original ash reference assembly was sequenced for the new version of the assembly, using Pacific Biosciences long-read sequencing and HiC performed by Cantata Bio LLC, a company providing genetic sequencing.
The updated reference genome has 2,024 scaffolds, compared with 89,514 in the previous assembly. Over 90% of the updated assembly is contained in 23 scaffolds that correspond to the 23 chromosomes of F. excelsior, indicating a very high level of completeness – the remaining 2,001 scaffolds only represent a small fraction of the genome.
This updated reference genome therefore allows analysis at the chromosomal scale rather than relying on small fragments. It will be a valuable resource for understanding the biology of ash, and in identifying variants within European ash that will contribute to efforts to improve the species’ resistance to pests and diseases, foremost of which is ash dieback, and to enhance its climate resilience.

A healthy ash tree
For scientists – you can find a link to the new Fraxinus excelsior reference genome here.
You can find out more about the ash pangenome work that Daniel is doing here and our pilot year project on ash genomics here.


